Tutorial#
The deepcell-types model predicts cell types from multiplexed spatial
proteomic images.
There are three main inputs to the cell type prediction pipeline:
The multiplexed image in channels-first format
A whole-cell segmentation mask for the image, and
A mapping of the channel index to the marker expression name.
Each of these components will be covered in further detail in this tutorial.
Example datasets#
This tutorial will make use of the spatial proteomic data available on the
HuBMAP data portal.
Users are encouraged to explore the portal for data of interest.
For convenience, a subset of the publicly-available spatial proteomic data
has been converted to a remote zarr archive.
The datasets in the zarr archive reflect the original HuBMAP indexing scheme
(i.e. HBM###_????_###, where # indicates a number nad ? indicates an
upper-case alphabetical character).
Interacting with the zarr hubmap data mirror requires a few additional dependencies:
pip install zarr\>2 s3fs rich
Note
The hubmap data mirror uses zarr format v3, thus requires zarr>2 to be
installed.
import zarr
if not zarr.__version__.startswith("3"):
raise EnvironmentError(
f"The tutorial requires `zarr>3`, version {zarr.__version__} found."
)
Exploring the archive#
z = zarr.open_group(
store="s3://deepcelltypes-demo-datasets/hubmap.zarr",
mode="r",
storage_options={
"anon": True,
"client_kwargs": dict(region_name="us-east-1"),
},
)
High-level structure of the data archive:
z.tree()
/ ├── HBM267_BZKT_867 │ ├── image (19, 9072, 9408) uint16 │ └── segmentations │ ├── cellsam (9072, 9408) uint32 │ └── torch_mesmer (9072, 9408) uint32 ├── HBM685_PCCJ_427 │ ├── image (54, 9510, 9993) uint16 │ └── segmentations │ ├── cellsam (9510, 9993) uint32 │ └── torch_mesmer (9510, 9993) uint32 └── HBM994_PDJN_987 ├── image (37, 2048, 2048) int16 └── segmentations ├── cellsam (2048, 2048) uint32 └── torch_mesmer (2048, 2048) uint32
A more detailed look at the datasets:
import pandas as pd # for nice html rendering
summary = pd.DataFrame.from_dict(
{k:
{
"tissue": z[k].attrs["tissue"],
"technology": z[k].attrs["modality"],
"Num ch.": z[k]["image"].shape[0],
"shape": z[k]["image"].shape[1:],
}
for k in z.group_keys()
},
orient="index",
)
summary.sort_index()
| tissue | technology | Num ch. | shape | |
|---|---|---|---|---|
| HBM267_BZKT_867 | spleen | codex | 19 | (9072, 9408) |
| HBM685_PCCJ_427 | intestine | codex | 54 | (9510, 9993) |
| HBM994_PDJN_987 | uterus | mibi | 37 | (2048, 2048) |
In the interest of minimizing network bandwidth, we’ll use the HBM994_PDJN_987
dataset to demonstrate the deepcell-types inference pipeline.
k = "HBM994_PDJN_987"
Dataset anatomy#
As noted above, the cell-type prediction pipeline requires the multiplexed image,
the channel-name mapping, and a segmentation mask for the image.
The multiplexed image is stored in the image array for each dataset, and the
channel mapping is stored under the key "channels" in the image metadata.
Note that these two inputs are derived directly from the corresponding datasets
on the HuBMAP data portal.
ds = z[k]
img = ds["image"][:] # Load data into memory
chnames = ds["image"].attrs.get("channels")
# Sanity check: ensure that channel name list is the same size as the number of
# channels in the image
len(chnames) == img.shape[0]
True
Another bit of metadata that is useful (when available) is the pixel size of the image, in microns-per-pixel. While not strictly required, this can improve predictions by tamping down variability in image scaling. This information is stored in the dataset metadata.
mpp = ds["image"].attrs["mpp"]
mpp
0.6
Running the cell-type prediction pipeline#
Note
Both cellSAM and deepcell-types models can in principle be run on CPUs, but
it is strongly recommended that users make use of GPU-capable machines when
running cell segmentation/cell-type prediction workflows.
The final input is a segmentation mask.
deepcell-types has been intentionally designed for flexibility on this front
to better integrate into existing spatial-omics workflows.
However, for convenience, several pre-computed segmentation masks are stored
in the data archive: one computed by Mesmer
(available at ds["segmentations/torch_mesmer"])
and a second by CellSAM
(available at ds["segmentations/cellsam"]).
In this tutorial, we will demonstrate how to use one of these models to construct a full cell-type inference pipeline.
Cell segmentation with cellSAM#
In order to use cellSAM, it must be installed in the environment, e.g.
pip install git+https://github.com/vanvalenlab/cellSAM.git
import numpy as np
from cellSAM.cellsam_pipeline import cellsam_pipeline
/home/administrator/repos/deepcell-types/dct13env/lib/python3.13/site-packages/cellSAM/model.py:12: UserWarning: pkg_resources is deprecated as an API. See https://setuptools.pypa.io/en/latest/pkg_resources.html. The pkg_resources package is slated for removal as early as 2025-11-30. Refrain from using this package or pin to Setuptools<81.
from pkg_resources import resource_filename
For convenience, channels corresponding to nuclear markers and a whole-cell marker are stored in the dataset metadata.
Note
Nuclear markers are typically unambiguous. The whole-cell channel selection
on the other hand is less well-defined.
Users are encouraged to try different channels or combinations of
channels for improved whole-cell segmentation results.
The membrane_channel selection in the metadata is arbitrary and provided
for convenience.
# Extract channels for segmentation
nuc, mem = ds.attrs["nuclear_channel"], ds.attrs["membrane_channel"]
im = np.stack(
[img[chnames.index(nuc)], img[chnames.index(mem)]],
axis=-1,
).squeeze()
CellSAM expects multiplexed data in a particular format. See the cellsam docs for details.
# Format for cellsam
seg_img = np.zeros((*im.shape[:-1], 3), dtype=im.dtype)
seg_img[..., 1:] = im
Finally, run the segmentation pipeline:
mask = cellsam_pipeline(
seg_img,
block_size=512,
low_contrast_enhancement=False,
use_wsi=True,
gauge_cell_size=False,
)
# Sanity check: the segmentation mask should have the same W, H dimensions as
# the input image
mask.shape == img.shape[1:]
True
Let’s perform a bit of post-processing to ensure that the segmentation mask (represented as a label image) is sequential.
import skimage
mask, _, _ = skimage.segmentation.relabel_sequential(mask)
mask = mask.astype(np.uint32)
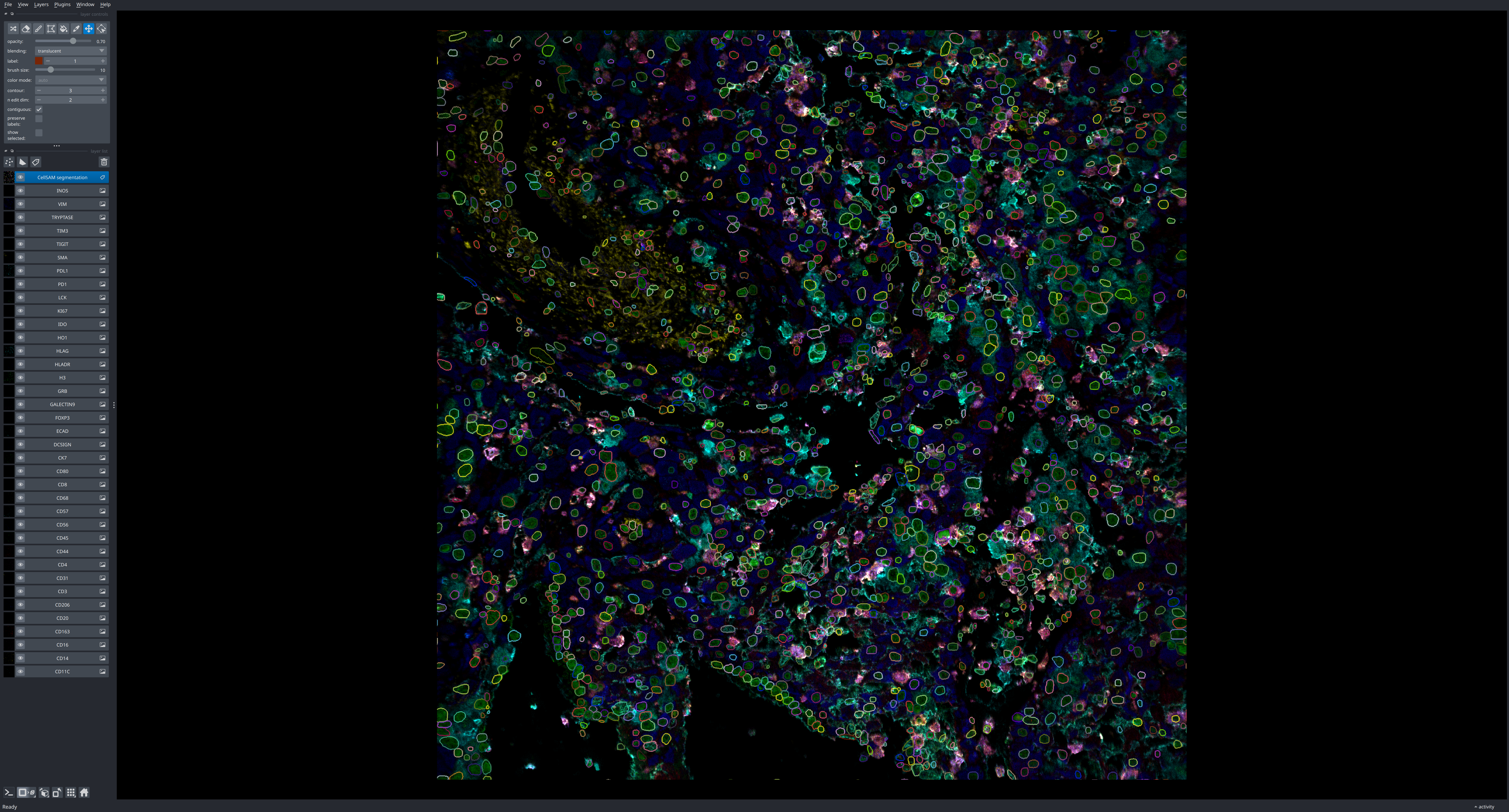
Visualizing results#
Note
Multiplexed images and their analysis products are extremely information dense; users are
strongly recommended to run tutorials locally to leverage napari for interactive
visualization.
import napari
nim = napari.Viewer(show=True) # Headless for CI; set show=True for interactive viz
# Compute contrast limits
cl = [(np.min(ch), np.max(ch)) for ch in img]
# Visualize multiplex image
nim.add_image(img, channel_axis=0, name=chnames, contrast_limits=cl);
# Add segmentation mask
mask_lyr = nim.add_labels(mask, name="CellSAM segmentation")
mask_lyr.contour = 3 # Relatively thick borders for static viz
Cell-type inference with deepcell-types#
We now have all the necessary components to run the cell-type inference pipeline.
import deepcell_types
To run the inference pipeline, you will need to download a trained model. See Models for details.
# Model & system-specific configuration
model = "deepcell-types_2025-06-09"
# NOTE: if you do not have a cuda-capable GPU, try "cpu"
device = "cuda:0"
# NOTE: For machines with many cores & large RAM (e.g. GPU nodes), consider
# increasing for better performance.
num_data_loader_threads = 1
With the system all configured, we can now run the pipeline:
cell_types = deepcell_types.predict(
img,
mask,
chnames,
mpp,
model_name=model,
device_num=device,
num_workers=num_data_loader_threads,
)
Predictions are provided in the form of list of strings, where the order of the list is given by the ordering of cell indices in the segmentation mask. Since we ordered the mask indices above, it’s straightforward to make this mapping explicit:
idx_to_pred = dict(enumerate(cell_types, start=1))
pd.DataFrame.from_dict( # For nice table rendering
idx_to_pred, orient="index", columns=["Cell type"]
)
| Cell type | |
|---|---|
| 1 | Myofibroblast |
| 2 | Myofibroblast |
| 3 | Endothelial |
| 4 | Stellate |
| 5 | Myofibroblast |
| ... | ... |
| 1642 | NK |
| 1643 | EVT |
| 1644 | Myofibroblast |
| 1645 | NK |
| 1646 | EVT |
1646 rows × 1 columns
Depending on the subsequent analysis you wish to perform, it may be convenient to group the cells by their predicted cell-type:
from collections import defaultdict
# Convert the 1-1 `cell: type` mapping to a 1-many `type: list-of-cells` mapping
labels_by_celltype = defaultdict(list)
for idx, ct in idx_to_pred.items():
labels_by_celltype[ct].append(idx)
Here’s the distribution of predicted cell types for this tissue:
from pprint import pprint
print(f"Total number of cells: {(num_cells := np.max(mask))}")
pprint(
{
k: f"{len(v)} ({100 * len(v) / num_cells:02.2f}%)"
for k, v in labels_by_celltype.items()
},
sort_dicts=False,
)
Total number of cells: 1646
{'Myofibroblast': '100 (6.08%)',
'Endothelial': '43 (2.61%)',
'Stellate': '9 (0.55%)',
'NK': '311 (18.89%)',
'Fibroblast': '340 (20.66%)',
'Macrophage': '191 (11.60%)',
'SmoothMuscle': '54 (3.28%)',
'NKT': '9 (0.55%)',
'EVT': '485 (29.47%)',
'Epithelial': '82 (4.98%)',
'Melanocyte': '4 (0.24%)',
'CD4T': '8 (0.49%)',
'CD8T': '4 (0.24%)',
'HSEC': '5 (0.30%)',
'Microglial': '1 (0.06%)'}
Visualizing the results#
There are many ways to visualize the cell-type prediction data, each with their own advantages and disadvantages. One way is to add an independent layer for each predicted cell type. The advantage of this approach is that individual layers can be toggled to focus on a particular cell type during interactive visualization.
# Regionprops to extract slices corresponding to each individual cell mask
props = skimage.measure.regionprops(mask)
prop_dict = {p.label: p for p in props}
# Create a binary mask layer for each celltype and populate it
# using the regionprops
for k, l in labels_by_celltype.items():
ctmask = np.zeros_like(mask, dtype=np.uint8)
for idx in l:
p = prop_dict[idx]
ctmask[p.slice][p.image] = 1
mask_lyr = nim.add_labels(ctmask, name=f"{k} ({len(l)})")
mask_lyr.colormap = napari.utils.DirectLabelColormap(
color_dict={None: (0, 0, 0), 1: np.random.rand(3)}
)
